 |
A 25-year-old male presented complaining of longstanding poor vision. He had no systemic, ocular or family history. Entering acuity was 20/25- OD and 20/25 OS. His extraocular muscles were full, and no afferent pupillary defect was present. On slit lamp examination in the deeper part of the stroma, translucent, scaly polygonal opacities were seen appearing centrally; however, the peripheral intervening tissue was clear. His manifest refraction OD was -0.25D and -0.25D OS. He was correctable to 20/20 in both eyes. Intraocular pressure was 15mm Hg OU. The rest of his ocular health was unremarkable. Upon questioning, the patient reported previous eye exams but no mention of irregular findings.
Given the bilateral and symmetric nature of this presentation, it was believed that this patient had a corneal dystrophy (CD). CDs are a rare group of genetic eye disorders and regularly present in early childhood. They are not associated with systemic or environmental factors, nor are they caused by inflammation, infection or trauma. Instead, they are caused by a genetic mutation. CDs are hereditary, bilateral and symmetric and are typically slow-progressing, starting early in life but may not become clinically apparent until later.1,2 Most CDs affect men and women equally, with the exception of Fuchs’ dystrophy, which affects more women.
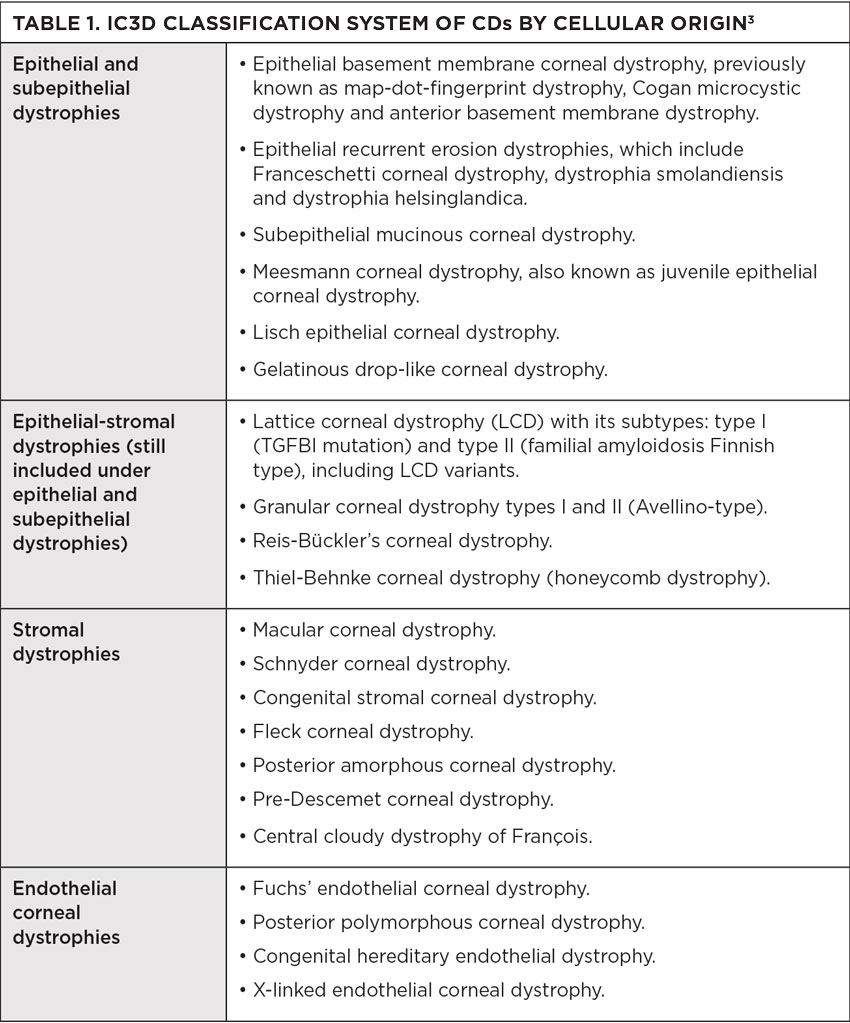
The cornea has five layers: the epithelium, Bowman’s membrane, the stroma, Descemet’s membrane and the endothelium. CDs are caused by a build-up of foreign material in one or more of these five layers. They are usually confined to one layer, thus most commonly classified by corneal location. More than 20 different CDs exist, and in 2008, the International Committee for Classification of Corneal Dystrophies (IC3D) was created to devise a current and accurate nomenclature for them. The IC3D classified CDs in order to reflect genetic, clinical and histological characteristics and created four categories.
• Category 1: A well-defined corneal dystrophy in which the gene has been mapped and identified with specific mutations known.
• Category 2: A well-defined corneal dystrophy that has been mapped to one or more specific chromosomal loci, but the genes remain to be identified.
• Category 3: A well-defined corneal dystrophy in which the disorder has not yet been mapped to a chromosomal locus.
• Category 4: Reserved for a suspected new or previously documented corneal dystrophy, although the evidence of it being a distinct entity is not yet convincing.
In 2015, the IC3D reviewed the new information on corneal dystrophies reviewed between 2008 and 2014. It proposed reclassification, that based categorization on cellular origin of dystrophies. This sorting uses the anatomical level affected, more accurately classifying the transforming growth factor beta-induced (TGFBI) dystrophies that affect multiple layers, instead of residing in one. With this classification, histopathologic and confocal images have been added to the templates. The IC3D found that confocal microscopy emerged as extremely helpful in discovering features of several CDs that previously required histopathologic examination. The dystrophies represent four categories: epithelial and subepithelial, epithelial-stromal, stromal and endothelial.1 This updated version has provided more clarity when diagnosing patients with a CD (Table 1).
Some of these dystrophies cause the cornea to lose transparency, which can cause blurred or lost vision. Other symptoms may include pain, tearing and foreign body sensation, but they can also present with none. Treatment of CDs ranges and is based on signs and symptoms. While some patients are asymptomatic, mild signs are often managed with early observation. If they progress, lubrication is recommended. Often, bandage lenses are used to help with recurrent erosions, while phototherapeutic keratectomy and corneal transplantation are reserved for persistent recurrent erosions and severely decreased vision.
 |
|
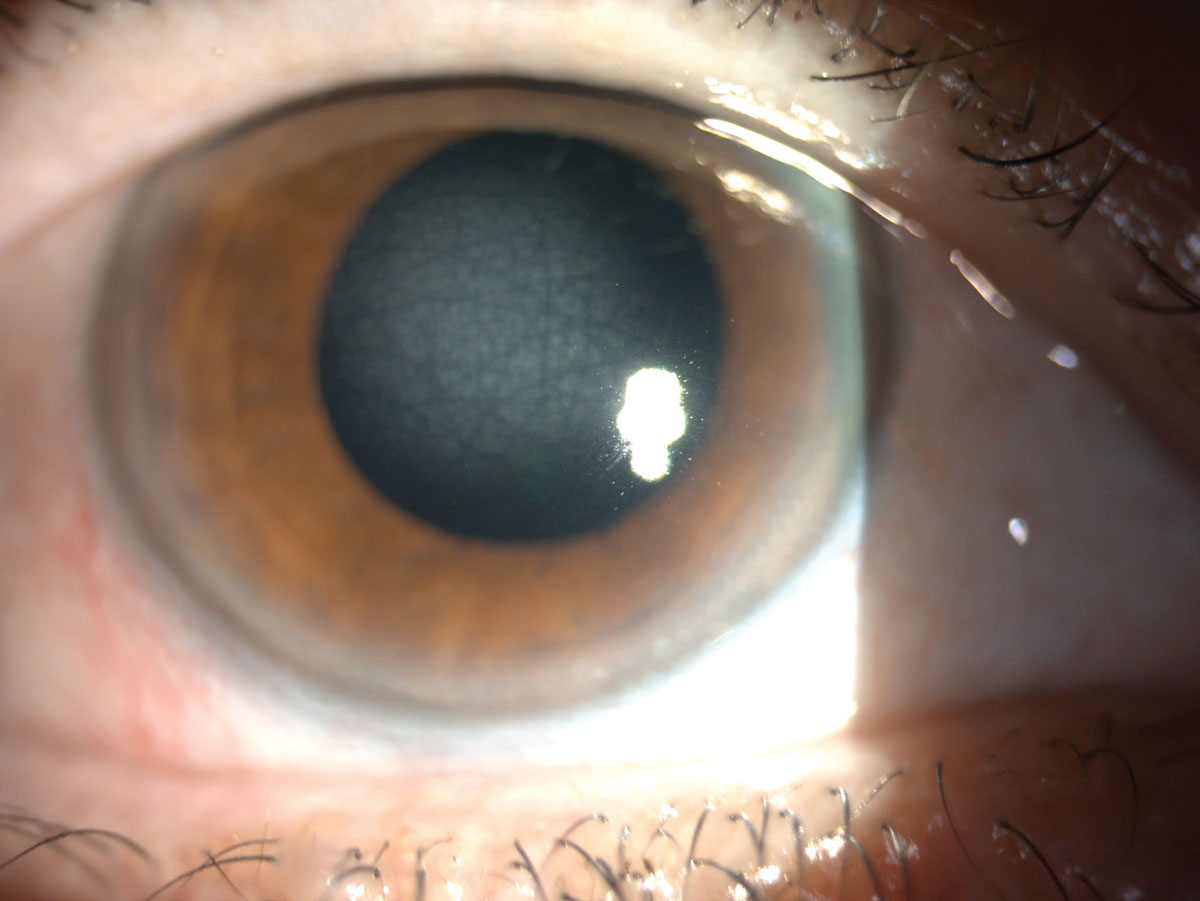
An example of central cloudy dystrophy of François. Click image to enlarge. |
Diagnosis
Our patient was diagnosed with a stromal CD, central corneal dystrophy of François (CCDF). CCDF possesses autosomal dominant inheritance with onset during the first or second decade of life.2 Often, it is described as appearing similar to crocodile shagreen.1 Signs of CCDF include translucent, scaly polygonal or rounded opacities residing deep within the stroma and are surrounded by clear tissue.4 It has also been described as a fluffy opacity, appearing as cracked ice. Pathologic findings include collagen of deep stroma, but anterior segment OCT findings are not conclusive at this time. Confocal microscopy in reported cases has revealed small, highly refractile granules and deposits throughout the stromal layers.5
Given that the stroma is a sizeable portion of the cornea, there is a large array of dystrophies affecting this area. The classification of stromal dystrophies was particularly affected in 2015 when the IC3D came out with new guidelines. The old classification had the following all listed as stromal dystrophies: lattice, granular, Avellino, macular, gelatinous drop-like, Schnyder, François-Neetans fleck and congenital hereditary corneal dystrophies. As the IC3D has learned more about these dystrophies’ cellular nature, the grouping was broken up and divided into the classification above.
CDs are mostly autosomal dominant diseases and present from the first to the third decade of life. Symptoms are initially not present; however, they can result in bilateral recurrent erosions, causing pain, tearing, foreign body sensation and eventually decreased vision. Signs are often recurrent erosions, bilateral corneal stromal deposits in various patterns and/or diffuse haze and for some, central corneal opacities or crystalline deposits with surrounding arcus. Treatment for stromal dystrophies often starts with observation and lubrication. In time, recurrent erosions can demand the need of bandage contact lenses and topical antibiotics. More severe cases can call for phototherapeutic keratectomy and even corneal transplantation. Even with corneal grafts, these dystrophies can reoccur.
 |
In day-to-day clinical care, a careful slit lamp examination with the use of optic section is recommended. Slit-lamp photography will also contribute significantly to documentation of and the ability to detect progression of CDs. The use of anterior segment OCT and confocal microscopy will, unquestionably, be a part of the future management of these conditions as more evidence is found.
1. Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies—edition 2. Cornea. 2015;34(2):117-59. 2. Cherny C, Sherman S. Detailing the dystrophies. RCCL. 2012;158(4):30-7. 3. Moshirfar M, Bennett P, Ronquillo Y. Corneal dystrophy. In: StatPearls. Treasure Island (FL): StatPearls Publishing. Updated August 7, 2023. 4. Lisch W, Weiss JS. Early and late clinical landmarks of corneal dystrophies. Exp Eye Res. 2020;198:108139. 5. Boyd K. What are corneal dystrophies. American Academy of Ophthalmology. Published September 28, 2022. aao.org/eye-health/diseases/corneal-dystrophies. Accessed August 18, 2023. |

