Corneal dystrophies are rare ocular disorders that often present in early childhood and arise independent of systemic and environmental factors; they are characterized by a hereditary, bilateral and symmetric nature.1,2 Dystrophies are typically slowly progressive and non-inflammatory, with some cases resulting in corneal opacification.1-3 Clinicians need to be able to distinguish these disorders from degenerations—changes from natural aging or disease that result in loss of normal corneal properties—although the distinction between the two is not always clear.2 Careful examination of every corneal layer in both eyes is required in order to confirm bilateral disease.2,4
Dystrophies are often confined within a specific layer and have traditionally been classified in this manner. More recently, it has been discovered that certain dystrophies may affect multiple layers simultaneously, resulting in clinicians using unique, characteristic landmarks to identify the individual dystrophies once they are fully developed.1,4
Here we dissect each corneal dystrophy, layer by layer, to aid clinicians in conducting a workup and arriving at a correct diagnosis.
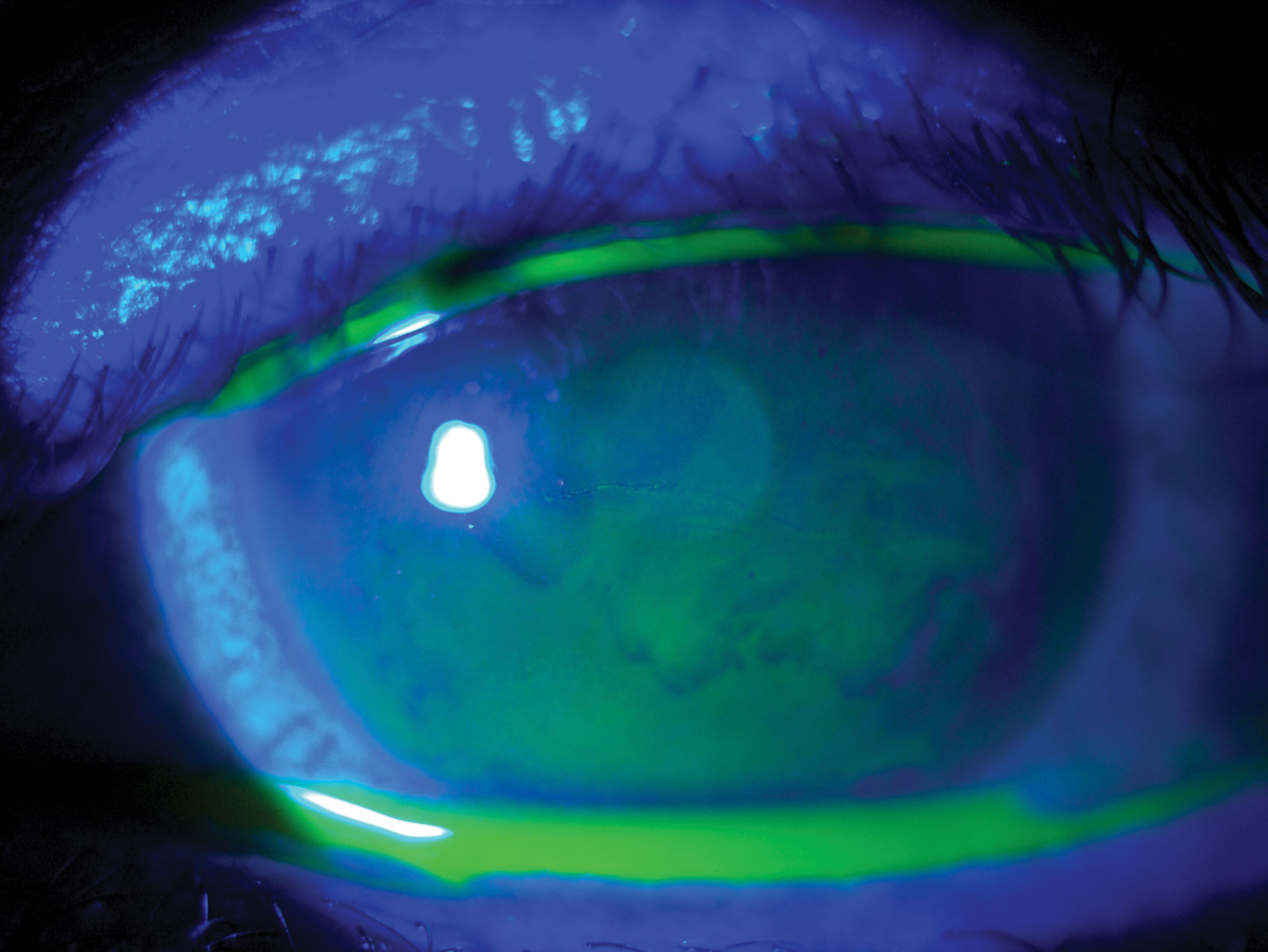 |
| Fig. 1. A resolving erosion due to epithelial basement membrane dystrophy. Click image to enlarge. |
Epithelial and Subepithelial Dystrophies
This category includes some of the more common forms. While troubling to the patient, they typically cause minimal vision loss.
Epithelial basement membrane dystrophy (EBMD). Also known as map-dot-fingerprint dystrophy, this condition is sometimes classified as a corneal degeneration due to minimal documented cases of autosomal dominant familial inheritance; many cases appear isolated or secondary to corneal trauma (Figure 1).1,2,4 EBMD occurs due to poor basal epithelial cell adhesion to the basal lamina, which predisposes the eye to recurrent corneal erosions (RCE).1
Corneal landmarks include grayish dots and lines in a characteristic pattern as well as cysts and blebs that may appear as thickened, hazy and gray epithelium with scalloped borders.1,2,4 While some patients may be asymptomatic, others can present with decreased visual acuity and/or monocular diplopia, as well as painful, recurring episodes of corneal erosion that may wake the patient.1,2
Initial RCE treatment is typically conservative, consisting of copious lubrication particularly during episodes of acute RCE.2 If corneal erosions persist or the dystrophy becomes visually significant, consider surgical RCE treatment options; these include phototherapeutic keratectomy (PTK) and anterior stromal puncture (ASP).2
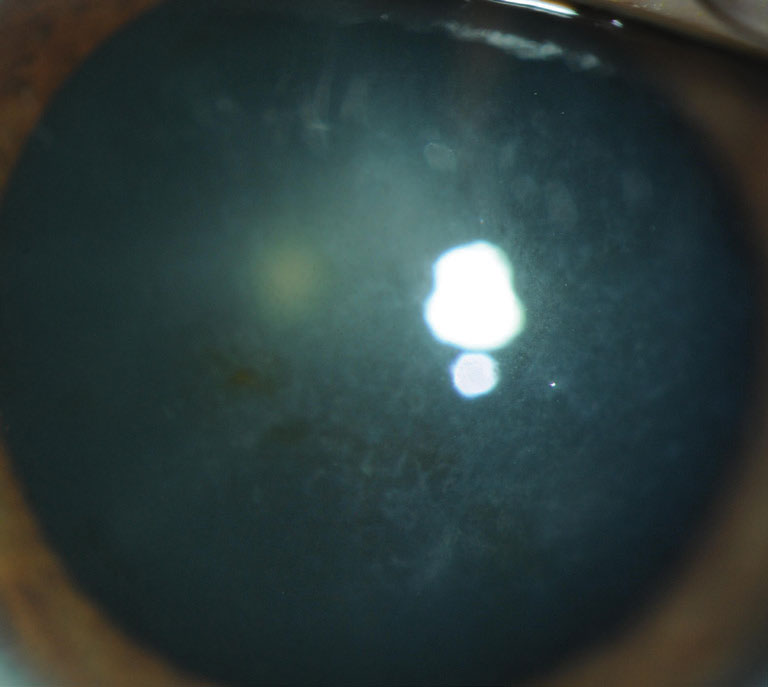 |
| Fig. 2. Signs of Reis-Bucklers corneal dystrophy include confluent geographic-like opacities at Bowman’s layer that become less discrete. Click image to enlarge. |
Epithelial recurrent erosion dystrophies (EREDS). These entail episodes of painful epithelial erosions lasting under one week, with erosive episodes often commence at night.1,2,4 Although the frequency of erosive episodes slowly diminishes with advancing age, it’s possible for diffuse, central subepithelial opacities to develop and impair visual acuity—with keloid formations developing in certain cases.1,2 Subepithelial pannus of the inferior cornea, intraepithelial bullae, amyloid deposition and microscopic gelatinous buds within the basal layers may be observed in EREDS variants.1,2,4
Conservative treatment of RCE may be sufficient during recurrent episodes; surgical RCE treatment, such as debridement or stromal puncture, is often necessary to address the fibrinous pannus and remove corneal opacities.2,4
Meesmann corneal dystrophy (MECD). This condition may follow a static or slowly progressive course resulting in moderately decreased in visual acuity.1,2,4 Signs of MECD include tiny, translucent intraepithelial vesicles extending from limbus to limbus that are more densely concentrated within the interpalpebral area, as well as diffuse gray opacities.1,2 Patients may be asymptomatic but can be symptomatic for decreased vision, glare, light sensitivity and foreign body sensation, with superficial punctate keratopathies or RCEs contributing to surface pain.1,2,4
Treatment consists of ocular lubrication, bandage contact lens wear in cases of epithelial defects or surgical RCE treatment in severe instances.2,4
Lisch epithelial corneal dystrophy (LECD). Characterized by clear microcysts as well as gray-white opacities in whorl, radial, band, feather and club-shaped patterns, LECD progresses slowly from periphery to the center, with an appearance similar to that of band keratopathy.1,2,4 Patients may be asymptomatic or present with decreased vision or photophobia.1,2,4 Treatment options include bandage contact lens wear, PTK and limbal stem cell transplantation.4
Gelatinous drop-like corneal dystrophy (GDLD). This progressive epithelial dystrophy displays a higher prevalence in the Japanese population.1,2,4 GDLD may present similarly to band keratopathy with multiple mulberry-like opacities that stain with fluorescein.1,2,4 Other signs include superficial vascularization, stromal opacification and large kumquat nodules.1,4 Patients may be symptomatic for decreased vision, photophobia, tearing, redness and foreign body sensation.1,2,4 Although surgical options are available, including superficial keratectomy, lamellar keratoplasty and penetrating keratoplasty, recurrence is common within a few years and may result in visual disability.1,2
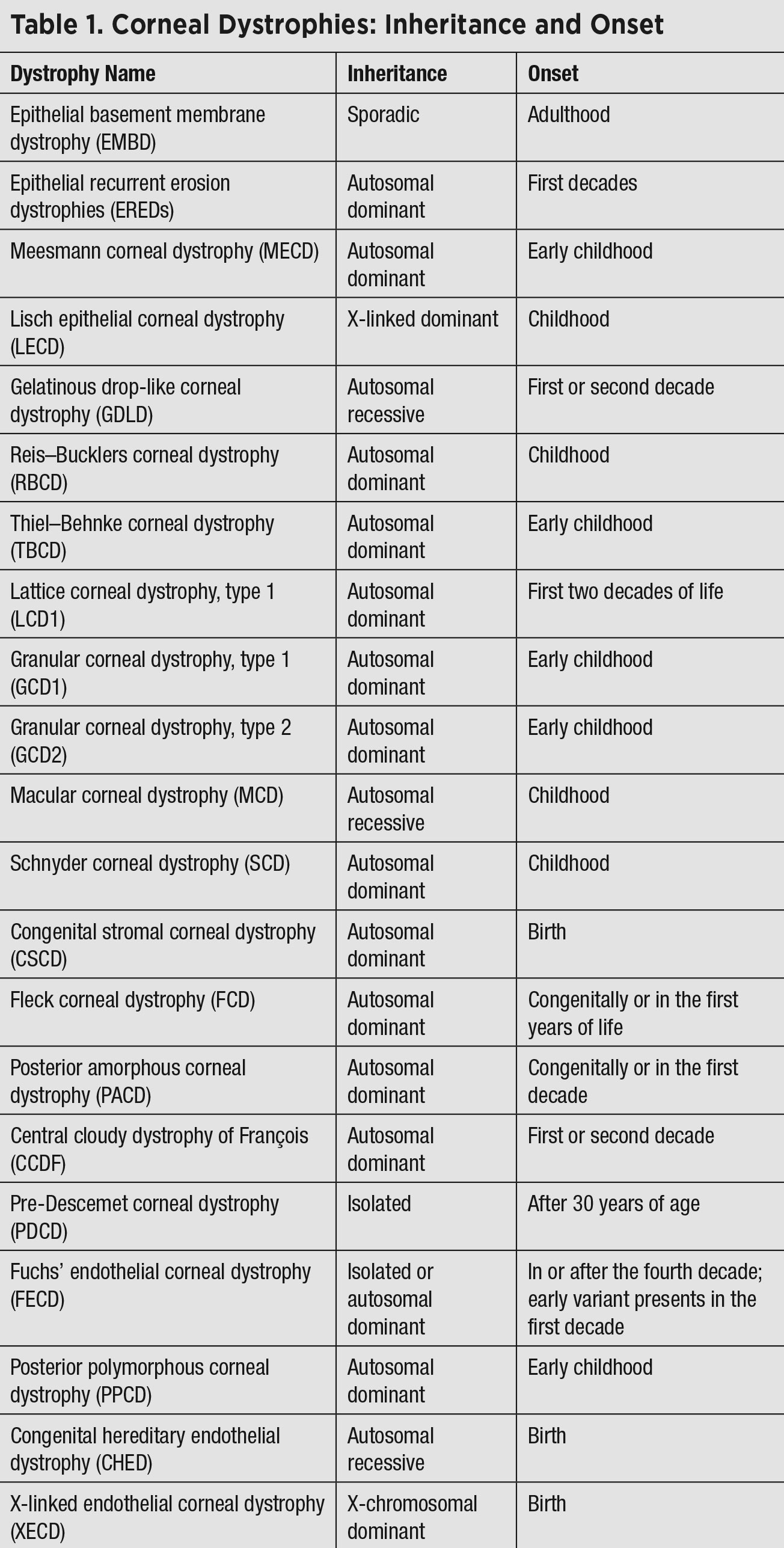 |
| Click table to enlarge. |
Epithelial-stromal TGFBI Dystrophies
Conditions in this category all arise from an autosomal-dominant mutation in the gene responsible for production of transforming growth factor β-induced (TGFBI), an extracellular matrix protein involved in cellular adhesion. Some of these disorders can be difficult to distinguish from one another, given their similar symptoms.
Reis–Bucklers corneal dystrophy (RBCD). This condition results in early visual impairment.1,2,4 Signs include confluent geographic-like opacities at Bowman’s layer that become less discrete and later extend to the limbus and deeper into the stroma1,2 (Figure 2). Vision slowly deteriorates and painful RCEs occur, although they diminish with time.1,2 RCEs are treated conservatively, while PTK and automated lamellar keratoplasty (ALK) are used to treat visual impairment. Recurrences are often seen after one year post-op.4
Thiel–Behnke corneal dystrophy (TBCD). Often difficult to distinguish from RBCD, this dystrophy’s signs include flecks or diffusely scattered irregular opacities at Bowman’s layer, with later symmetrical subepithelial honeycomb opacities.1,4 Although the opacities are often central, they can progress to the corneal periphery and deeper into the stroma with age.1 Patients may be symptomatic for RCEs during earlier decades with slowly progressive visual impairment due to scarring.1,2 Treatment includes lubrication, PTK or ALK, although there is a risk for recurrence.4
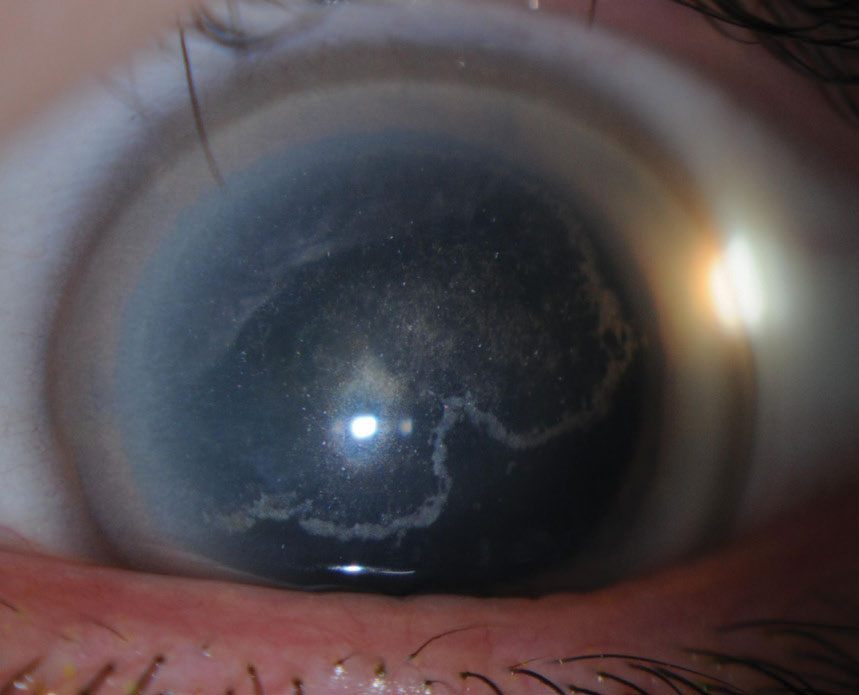 |
| Fig. 3. Signs of Schnyder corneal dystrophy include decreased photopic visual acuity, glare and decreased corneal sensitivity. Click image to enlarge. |
Lattice corneal dystrophy, type 1 (LCD1). Amyloid deposition within basement membranes in LCD1 patients gives rise, early on, to superficial central fleck opacities and deeper fine, translucent lattice lines within the superficial stroma. These opacities later spread throughout peripheral cornea and within deeper layers, avoiding far peripheral stroma.1,2 The deposits contribute to neuropathy and lead to corneal hypoesthesia, while RCEs also occur and contribute to pain and visual impairment.1,2
By the fourth decade, patients are often symptomatic with significant visual impairment.1,2 Deposition within the trabecular meshwork may contribute to ocular hypertension and glaucoma, while deposition within other organs can lead to numerous systemic complications.2 Treatment involves PTK, DALK or PKP, although deposits may recur post-op; intraocular pressure (IOP) must continue to be monitored.1,2,4
Granular corneal dystrophy, type 1 (GCD1). Hyaline deposition produces a radiating vortex pattern superficial to Bowman’s layer in these patients.1,4 Over time, dense crumb-like and snowflake-like granules arise but do not extend to the limbus, although they may extend to the deeper stroma.1,4 Symptoms include glare, photophobia and severely decreased vision; RCEs may transpire.1,4 Treatment includes conservative and/or surgical RCE treatment, although the deposits may recur.4
Granular corneal dystrophy, type 2 (GCD2). Also known as Avellino dystrophy, this condition presents with subtle, superficial stromal white dots due to hyaline and amyloid deposition; these progress to dense superficial patches with moth-eaten centers that may appear star- or ring-shaped and accompany branching stromal opacities.1,4 Patients experience RCEs and progressively decreased vision with age.1,4 Therapy includes conservative RCE treatment or PTK, although recurrences may take place.4
 |
| Fig. 4. Schnyder corneal dystrophy as seen on retroillumination. Click image to enlarge. |
Stromal Dystrophies
Given that the stroma comprises the lion’s share of the cornea, there is a wide array of dystrophies affecting this layer. Systemic associations are possible—be sure to investigate any risk factors noted in the medical history.
Macular corneal dystrophy (MCD). Keratin sulfate deposition leads to the development of superficial, central, white snowflake opacities and fog involving the limbus and deep stroma in these patients.1,2,4 With time, corneal sensitivity diminishes, diffuse stromal haze evolves and Descemet’s membrane grays and develops guttae.1,2 Patients may develop RCEs and photophobia, with vision diminishing severely by the second decade.1,2,4 Lamellar keratoplasty is often required to treat deep stromal deposition, although PKP is indicated if there is endothelial involvement; recurrences may be observed.2,4
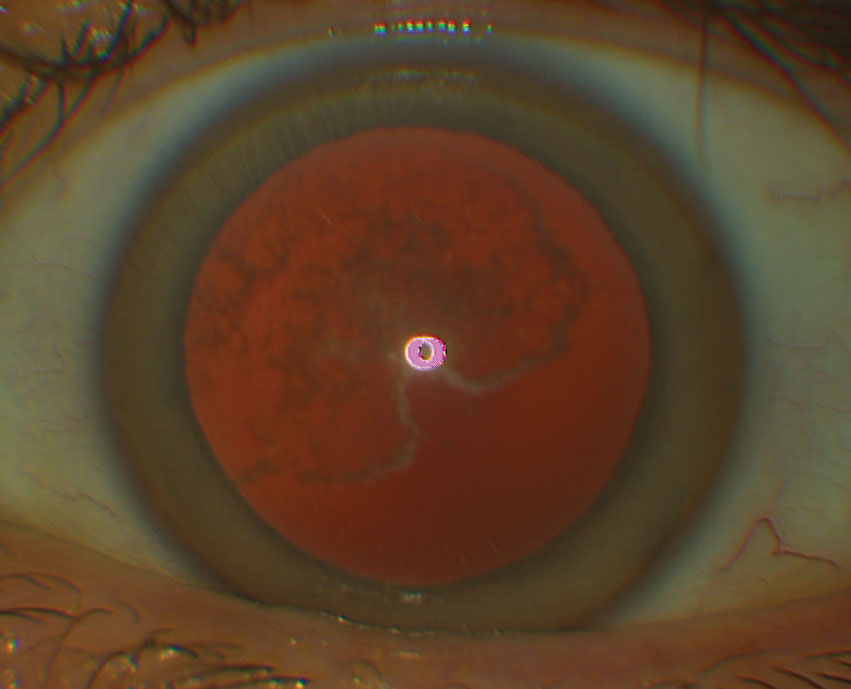
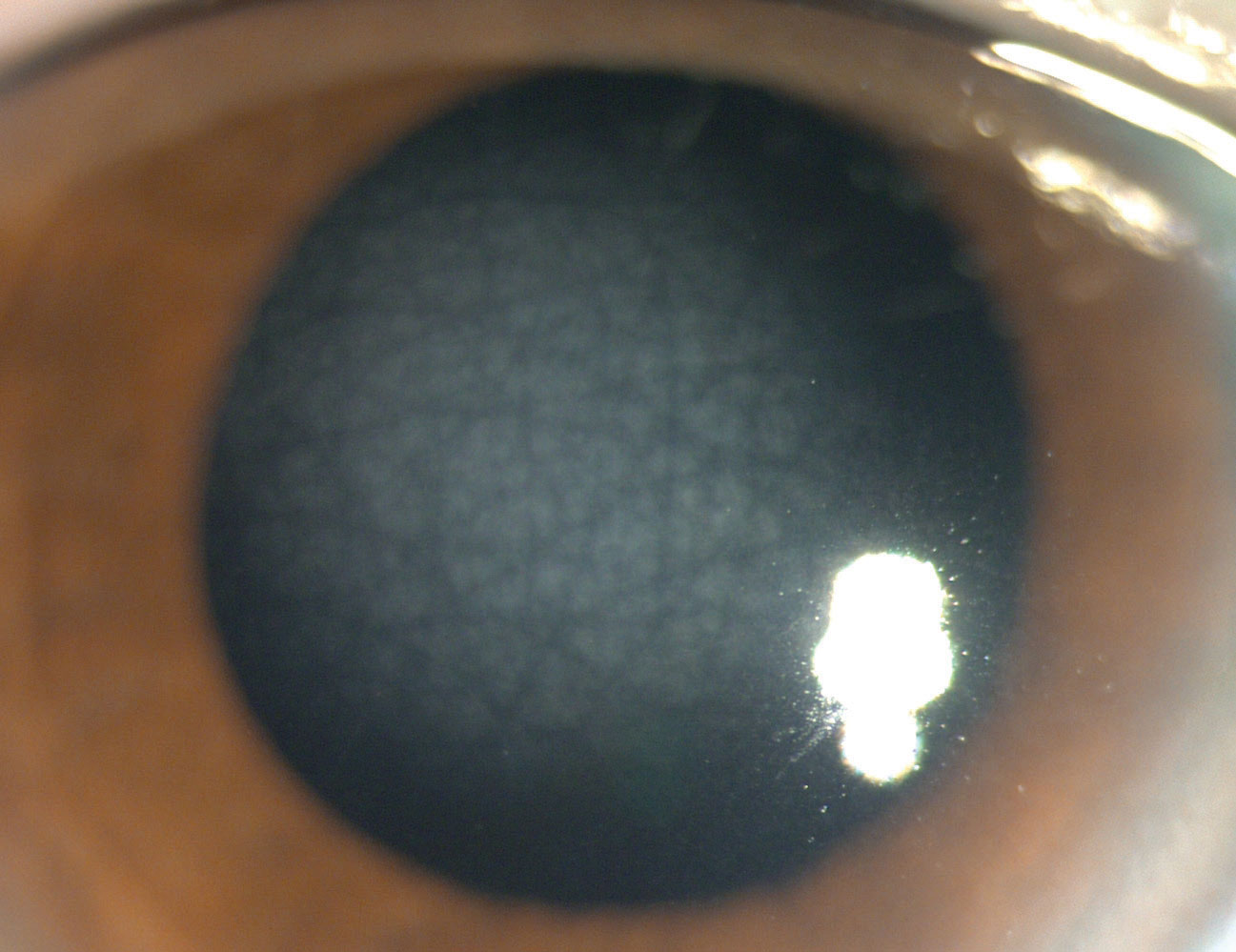
Schnyder corneal dystrophy (SCD). Although this condition often begins in childhood, diagnosis is usually made by the third decade, with identification further delayed in crystalline phenotypes, which comprise 50% of cases (Figures 3 and 4).1 Cholesterol is deposited within the corneal layers and leads to ring-like central opacities, comma-shaped subepithelial crystals, arcus lipoides and, with age, midperipheral stromal haze.1 Patients are symptomatic for decreased photopic visual acuity, glare and decreased corneal sensitivity, and may have associated dyslipidemia and xanthelasma.1,2
Ocular treatment includes PTK, deep ALK (DALK) or penetrating keratoplasty (PKP), and systemic cholesterol-lowering agents are often indicated.1,2
Congenital stromal corneal dystrophy (CSCD). Over a slowly progressive course, these patients will demonstrate bilateral diffuse corneal clouding with densely scattered white stromal opacities throughout the entire cornea.1,2,4 Patients may be symptomatic for photophobia and severe vision loss, which may be accompanied by strabismus.1,2
Ocular treatment options for this dystrophy include DALK and PKP, as well as treatment for amblyopia and strabismus. Systemic associations include type 1 diabetes or slowly progressive chronic renal involvement, which necessitates evaluation of blood glucose and renal function.2
 |
| Click table to enlarge. |
Fleck corneal dystrophy (FCD). This non-progressive disorder consists of tiny translucent discoid opacities or white stromal flecks, which may extend to the limbus.1,2,4 Patients may be asymptomatic, although mild photophobia and diminished corneal sensitivity may occur.1 Due to stable visual acuity, treatment is seldom indicated.2
Posterior amorphous corneal dystrophy (PACD). This condition presents as diffuse white, sheet-like opacities of the posterior stroma that may extend peripherally and to the limbus.1,4 Decreased corneal thickness, corneal flattening and hyperopia are often associated, as well as cornea plana.1,2 Additional reported associations include scleralization of the peripheral cornea, iris malformations, prominent Schwalbe’s line, iris processes, pupillary remnants, irido-corneal adhesions, corectopia and pseudopolycoria, although there is no association with glaucoma.1,2
Patients may be asymptomatic or experience mild photophobia and impairment of visual acuity, and the disorder is stable or slowly progressive.1,2 Treatments include PKP and DALK.2
Central cloudy dystrophy of François (CCDF). Presentation here resembles crocodile shagreen (a degeneration) within the anterior central cornea (Figure 5).1,2,4 Signs include translucent, scaly polygonal opacities within the deep stroma surrounded by clear intervening tissue.1,2 Treatment is seldom indicated for CCDF, as patients are asymptomatic and the course is nonprogressive.1,2
Pre-Descemet corneal dystrophy (PDCD). This generally presents with punctiform, focal gray opacities in the deep stroma anterior to Descemet’s membrane, which may resemble corneal farinata.1,2,4 Many forms of PDCD are nonprogressive and patients remain asymptomatic, with no indicated treatment.1 Due to common systemic association with ichthyosis, dermatologic work-up may be warranted.2
 |
| Fig 5. The appearance of central cloudy dystrophy resembles that of crocodile shagreen within the anterior central cornea. Click image to enlarge. |
Endothelial Dystrophies
These disorders, characterized by opacities or lesions, can threaten corneal function as well as vision, given their location.
Fuchs’ endothelial corneal dystrophy (FECD). The most prevalent of this category of dystrophies, this condition displays a female predominance and presents with characteristic corneal guttae—wart-like deposits on the inner aspect of Descemet’s membrane resembling beaten metal that start centrally and disseminate peripherally.1,2 In later stages, endothelial decompensation occurs with subsequent stromal edema, epithelial bullae, subepithelial fibrosis and superficial vascularization.1,2,4
Patients are intermittently symptomatic for decreased vision during episodes of epithelial and stromal edema, which is often worse upon waking, and vision may progressively deteriorate with time.1,2,4 Bullous keratopathy may cause pain, photophobia and epiphora, with ruptured bullae resulting in epithelial erosions.2,4
Treatment involves hyperosmolar agents in the edematous stage to promote stromal deturgescence, bandage contact lens wear to control pain in the bullous keratopathy stage and surgical replacement of the endothelium in severe disease with Descemet’s membrane endothelial keratoplasty (DMEK), posterior lamellar keratoplasty (typically Descemet’s stripping automated endothelial keratoplasty) or PKP.2,4
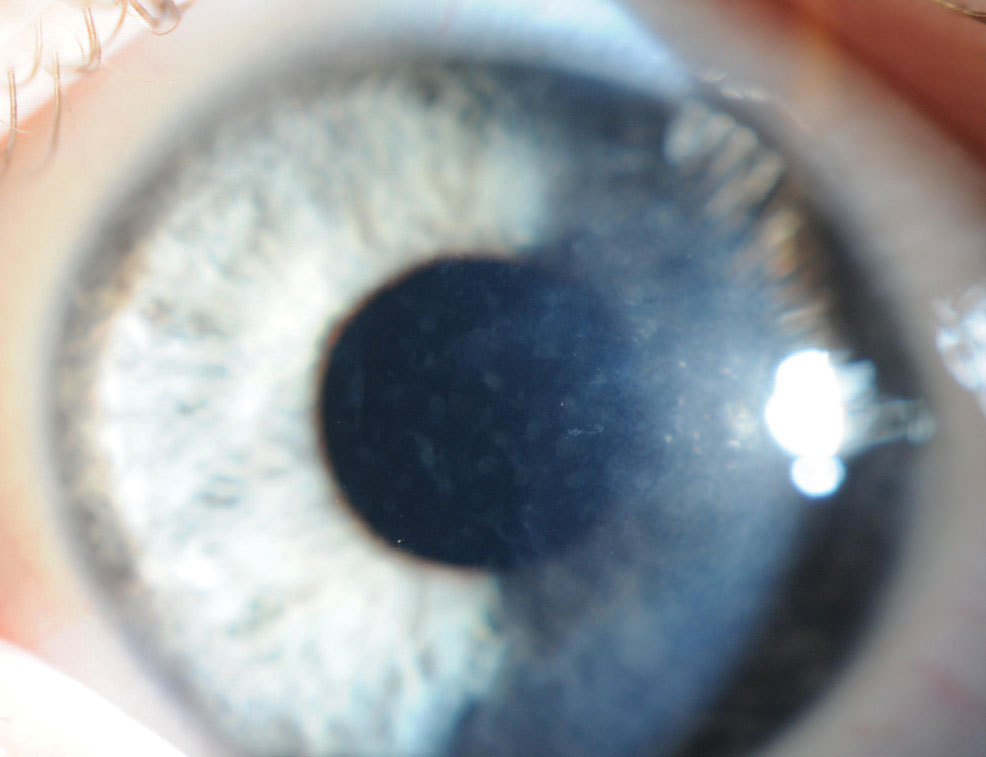
Posterior polymorphous corneal dystrophy (PPCD). This condition begins with asymmetric, geographic gray opacities of Descemet’s membrane and endothelium, as well as vesicular zones surrounded by gray circular opacities and/or white bands of flaky material resembling railroad tracks (Figure 6).1,2,4 It’s common for patients to develop corneal edema with subsequent visual impairment, which can necessitate keratoplasty.1 Peripheral iridocorneal adhesions exist in 25% of cases and may lead to elevated IOP.1,4 Patients may be symptomatic for foreign body sensation, decreased vision or photophobia.2 Systemic associations include herniation and warrant a gastrointestinal work-up.2
 |
| Fig. 6. Signs of posterior amorphous corneal dystrophy start out with asymmetric gray opacities of Descemet’s membrane and endothelium. Click image to enlarge. |
Congenital hereditary endothelial dystrophy (CHED). Developing in the fifth month of gestation, with a generally nonprogressive course thereafter, CHED leads to a decrease in endothelial cell count with dysfunction.2 The cornea appears opacified with a diffused, bluish edema and warrants differentiation from congenital glaucoma.2,4 Additional clinical signs include a thickened Descemet’s membrane, pseudo-bullous keratopathy, corneal thickening and nystagmus.2
CHED patients are typically symptomatic for severe visual loss and may be treated with hyperosmolar drops for edema or surgical endothelium replacement in severe disease.2,4 Systemic association includes neurosensory hearing loss, and audiology/ENT work-up is warranted.2
X-linked endothelial corneal dystrophy (XECD). This condition presents in males with congenital corneal clouding wherein the cornea may be diffusely hazy or milky-white in appearance.1 Additional clinical signs include moon crater–like endothelial changes with possible secondary subepithelial band keratopathy and nystagmus.2,4 Patients are symptomatic for decreased vision, although the course is minimally progressive.1
PTK can be performed in certain cases of subepithelial band keratopathy, while surgical endothelial replacement is performed in visually significant congenital haze.4
Is There a Role For Genetic Testing?With the advent of genotyping, the molecular genetic basis of numerous corneal dystrophies has been described and we are now able to genetically screen for certain suspected corneal dystrophies and confirm diagnosis.17 Online-based genetic testing services allow the clinician to order gene panels specifically for corneal dystrophies, although limited information exists regarding the quality or utility of the sequencing information. The long-term goal of genetic testing is to employ this knowledge for the development of targeted therapeutic approaches by means of gene therapy.17,19 The cornea may be an appealing target for gene therapy due to easy access, corneal immune privilege and often monogenic or Mendelian inheritance of corneal dystrophies.18 However, to this day, most corneal gene therapy studies have been conducted only in animal models or in vitro.18 Additionally, the mutational heterogeneity inherent to corneal dystrophies presents an obstacle to gene therapy development, which generally targets a single, primary gene defect.19 This compels further research into mutation-independent approaches, such as by means of RNA interference followed by gene replacement.19 Therefore, although extensive research is being performed to develop a targeted treatment for corneal dystrophies and circumvent surgical therapy, this is not the standard of care at this time. |
Management
Many corneal dystrophies produce pain and vision loss by means of RCE, and preventative measures such as topical lubrication and hypertonic saline are often necessary to protect the epithelium from erosion and bacterial infection.3 During acute RCE episodes, bandage contact lenses may be used along with cycloplegics and antibiotic ointment.3 Autologous serum drops, oral doxycycline, topical steroids, superficial keratectomy, PTK and ASP may be considered for recalcitrant RCEs.3,5 Keratoplasty may be used in extreme cases, although deposits may recur post-graft, particularly in stromal dystrophies.3,6 In certain cases, visual improvement may be achieved with the use of rigid gas permeable contact lenses.
Given the spectrum of visual disability that corneal dystrophies may cause and the possible need for surgical intervention, interdisciplinary care with cornea specialists is imperative, and systemic associations need to be evaluated by the respective specialties. Furthermore, examination of family members may be recommended to establish inheritance pattern and solidify diagnoses.
And now you’re up to speed with every corneal dystrophy! Although dystrophies are rare and sometimes challenging to diagnose, doing so can make a big difference with your patients.
Dr. Cherny received her doctor of optometry degree at SUNY and is currently pursing her contact lens/cornea and ocular disease residency at Mass Eye and Ear. She has no financial interests to disclose.
Dr. Sherman is an assistant professor of optometric sciences (in ophthalmology) and the director of optometry at Columbia University Medical Center. She has no financial interests to disclose.
1. Weiss JS, Moller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-edition 2. Cornea. 2015;34(2):117-59. 2. Bourges JL. Corneal dystrophies. J Fr Ophtalmol. 2017;40(6):e177-e92. 3. Omari AA, Mian SI. Recurrent corneal erosions in corneal dystrophies: a review of the pathogenesis, differential diagnosis, and therapy. Klin Monbl Augenheilkd. 2018;235(6):689-96. 4. Lisch W, Weiss JS. Early and late clinical landmarks of corneal dystrophies. Exp Eye Res. 2020;198:108139. 5. Eung Kweon K. PTK in Corneal Dystrophy. Cornea. 2004;23(4):323–4. 6. Marcon AS, Cohen EJ, Rapuano CJ, et al. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003; 22(1):19-21. 7. Venkateswaran N, Galor A, Wang J, et al. Optical coherence tomography for ocular surface and corneal diseases: a review. Eye and Vision. June 12, 2018. 8. Elhardt C, Priglinger SG, Karakolova Y, et al. Corneal dystrophies in optical coherence tomography. Ophthalmologe. 2019;116(9):857-64. 9. De Azavedo Magalhães, O, Rymer S, Marinho DR, et al. Optical coherence tomography image in gelatinous drop-like corneal dystrophy: case report. Arq Bras Oftalmol. 2012;75(5):356-7. 10. Perez M, Joubert R, Chiambaretta F. In vitro histological analysis, in vivo confocal microscopy and anterior segment spectral domain OCT in a case of Lisch epithelial corneal dystrophy. J Fr Ophtalmol. 2019;42(8):e363-6. 11. Ghazal W, Georgeon C, Grieve K, et al. Multimodal imaging features of schnyder corneal dystrophy. J Ophthalmol. 2020;2020:6701816. 12. Cervantes AE, Gee KM, Whiting MF, et al. Confirmation and refinement of the heterozygous deletion of the small leucine-rich proteoglycans associated with posterior amorphous corneal dystrophy. Ophthalmic Genet. 2018;39(4):419-24. 13. Anuradha R, Dhasmana R, H Bahadur. Unilateral lattice corneal dystrophy in a young female: a unique case report. Nepal J Ophthalmol. 2017;9(18):66-9. 14. Wertheimer CM, Elhardt C, Wartak A, et al. Corneal optical density in Fuchs endothelial dystrophy determined by anterior segment optical coherence tomography. Eur J Ophthalmol. 2020;31(4):1771-8. 15. Guier CP, Patel BC, Stokkermans TJ, Gulani AC. Posterior polymorphous corneal dystrophy. StatPearls. August 11, 2021. www.ncbi.nlm.nih.gov/books/NBK430880/. Accessed August 23, 2021. 16. Fogla, R. Role of anterior segment OCT for descemet membrane stripping during descemet membrane endothelial keratoplasty in eyes with congenital hereditary endothelial dystrophy. Cornea. 2021;40(4):458-61. 17. Aldave AJ, Sonmez B. Elucidating the molecular genetic basis of the corneal dystrophies: are we there yet? Arch Ophthalmol. 2007;125(2):177-86. 18. Amador C, Shah R, Ghiam S, Kramerov AA, Ljubimov AV. Gene therapy in the anterior eye segment. Curr Gene Ther. April 22, 2021. [Epub ahead of print.] 19. Lakshminarayanan R, Chaurasia SS, Anandalakshmi V, Chai SM, et al. Clinical and genetic aspects of the TGFBI-associated corneal dystrophies. Ocul Surf. 2014;12(4):234-51. |

